如何在云计算平台上完成分子对接
 2022年12月6日 11:57
2022年12月6日 11:57分子对接的基本原理就是把配体分子放在受体活性位点的位置,然后按照几何互补和能量互补的原则来实时评价配体与受体相互作用的优劣,最终找到这两个分子之间最佳的结合模式。
分子对接分为刚性、半柔性和柔性对接。不同的对接软件又可以分为:商业软件和学术软件,而分子对接的计算结果,则体现为打分函数的不同。目前用的比较多的分子对接软件有:AutoDock、AutoDock Vina、LeDock、rDock,这些都是学术软件;商业软件有:Glide、GOLD、MOE Dock、Surflex-Dock、LigandFit、FlexX等等。
做分子对接,一般对接的分子量都很大,到底有多大?就是很大!
我来举个例子,通常分子对接的采样,都是百万到千万级别的分子
而事实上可用于药物发现的有机分子有多少?
超过10的60次方!
我们取的只是沧海一粟罢了。
这么大的对接量,对算力的需求肯定少不了呀!
那怎么办呢?
具体,我以AutoDock-Vina分子对接为例。
AutoDock-Vina 是用于分子对接和虚拟筛选的开源程序,由Scripps研究所分子图形实验室的Oleg Trott博士设计和实现,是目前使用最为广泛的分子对接软件之一。此外,Vina可以利用系统上的多个CPU或CPU内核来显著缩短运行时间;北鯤云超算平台提供了丰富的cpu,gpu资源,此次教程以STAT3靶点的晶体结构与其原配体为例进行分子对接的详细步骤说明,就在北鯤云上演示。
准备工作
蛋白晶体结构由PDB数据库(http://www.rcsb.org/)下载,PDB编号为6NJS。首先需要通过其他软件如Pymol、Glide、DS等去除蛋白中的水分子,删除多余的链,并把原配体分子提离出来。单独的蛋白文件和配体文件均保存为.pdb格式。
设置工作目录:
为了计算的方便以及后续文件的方便查找,我们首先设置一个工作目录,要注意文件路径需全为英文,否则会导致程序出现错误。
我这里在C盘中设立文件夹AutoDock,以后的计算均可保存在该文件夹中。
为了将不同的计算结果分开,我们再创建一个文件夹6NJS在AutoDock文件夹下。
AutoDock vina的计算需要用到两个程序,即vina.exe和vina_split.exe。其中vina.exe是用来做对接,vina_split.exe用来分割对接构象结果。
在运算之前,从下载好的文件夹中复制这两个程序至6NJS文件夹中。同时将保存的蛋白及原配体的.PDB文件也放置在该文件夹中,其中6njs.pdb为蛋白文件,KQV701.pdb为原配体文件。
受体和配体的预处理
打开预装好AutoDockTools的windows工作站,在菜单栏点击File>ReadMolecule打开6NJS文件夹中的6njs.pdb。
-受体准备-
①除水:在准备工作中蛋白中水分子已被删除,这一步即可省略。
②加氢:Edit>Hydrogens>Add>OK
③计算电荷:Edit>Charges>ComputeGasteiger
④添加原子类型:Edit->Atoms->AssignAD4 type
⑤保存为.pdbqt文件:File->Save->WritePDBQT,此时可在6NJS文件夹下看到多了一个“6njs.pdbqt”文件
-配体准备-
点击鼠标右键>delete 在Dashboard中把受体分子删除。通过ADT菜单栏Ligand->Input->Open打开KQV701.pdb文件,弹出一个对话框,点击确定
①调整电荷:弹出窗口提示配体分子的总电荷数不是整数。点击Edit->Charges->Check Totals on Residues>Spread ChargeDeficit over all atoms in residue>Dismiss
②判定配体的root:ADT菜单栏Ligand>Torsion Tree>DetectRoot
③选择配体可扭转的键:Ligand>TorsionTree>Choose Torsions>Done,表示该分子32个键中有13个可旋转的。
其中红色表示不可旋转的键,绿色表示可旋转键,紫色表示不可扭转,通常为肽键。如果要设置某个键不可扭转,那么先按住shift键,然后鼠标单击即可(颜色变成紫色)。
④保存为.pdbqt文件:ADT菜单栏Ligand->Output->Saveas PDBQT,此时可在6NJS文件夹下看到多了一个“KQV701.pdbqt”文件
创建对接信息文件
在6NJS文件夹中创建一个配置文件:6njs.conf,这个文件里面写上用于对接的详细参数:
receptor = 6njs.pdbqt
ligand = KQV701.pdbqt
center_x = 13.24
center_y = 54.43
center_z = 0.27
size_x = 20.6
size_y = 31.1
size_z = 23.1
energy_range = 4
exhaustiveness = 12
num_modes = 10
receptor:表示受体分子的路径
ligand:表示配体分子的路径
center_x,center_y,center_z:表示活性位点盒子中心的坐标,我们这里以配体扩张法定义对接盒子,即以原配体所在位置为中心向外扩张一定的范围。如果没有原配体可以通过文献查找或工具预测的方法获得。
size_x,size_y,size_z:指定对接盒子的大小。这里设置的大小至少要包裹活性位点的空腔,但不宜设置过大,负责对接结果不准确,具体的设置可根据对接结果的好坏重新调整。
energy_range:与最优结合模型相差的最大能量值,单位是kcal/mol。我们设置为4则表示vina最多计算到与最优模型的能量差值为4就终止计算了。
exhaustiveness:用来控制对接的细致程度,默认值是8,设置值与电脑的配置相关,影响计算时间。
num_modes:最多生成多少个模型。此时,6NJS文件夹中应该包括配体分子,受体分子,参数文件,vina程序共7个文件。
执行计算
打开北鲲云超算平台,进入控制台
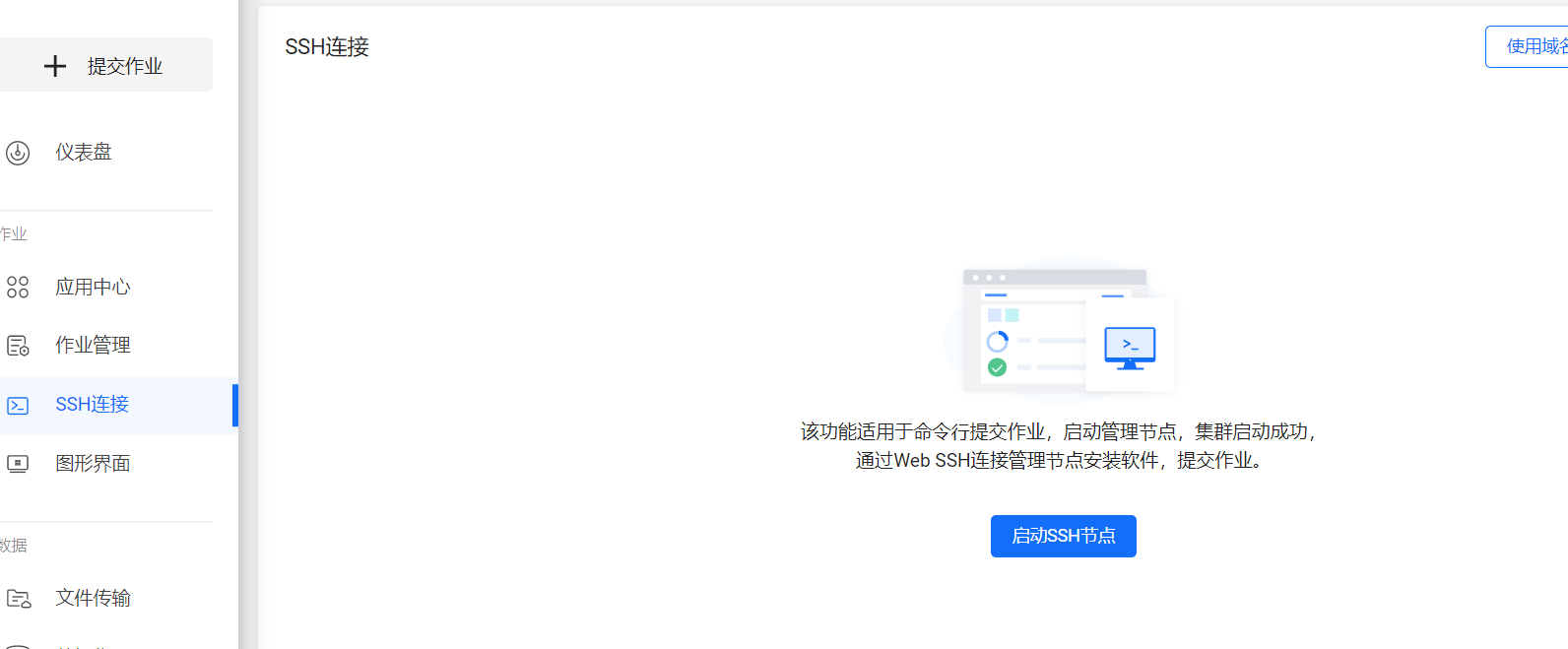
这里选择SSH连接,通过SSH链接启动一个管理节点,并连接进入管理节点。
通过win+R进入运行窗口,输入“cmd”进入命令行窗口,此时默认文件夹一般为C盘,
再输入
CD C:\AutoDock\6NJS
回车,进入该文件夹。
输入
vina--config 6njs.conf
回车,即可执行分子对接计算。
等待计算完成,一共得到10个模型结果,包括对接结合能分数,RMSD值,我们可以看到有多个结果的RMSD都小于2埃,说明本次分子对接结果还是比较可靠的。
在北鲲云超算平台完成分子对接计算的过程还是比较简单的,只要按照上述步骤即可完成,中途如果遇到问题可以随机联系我们的技术支持,技术支持随时在线解决大家的疑惑。
除了简便的操作之外,在平台上还有海量资源供大家选择,不用担心要排队或者ddl赶不上啦!





















工程师必备
- 项目客服
- 培训客服
- 平台客服
TOP




