GORMACS如何使用?一个方法快速完成动力学模拟计算
 2022年11月14日 15:26
2022年11月14日 15:26GROMACS是一个功能强大的分子动力学的模拟软件,其在模拟大量分子系统的牛顿运动方面具有极大的优势。
它可以用分子动力学、随机动力学或者路径积分方法模拟溶液或晶体中的任意分子,进行分子能量的最小化,分析构象等。它的模拟程序包包含GROMACS力场(蛋白质、核苷酸、糖等),研究的范围可以包括玻璃和液晶、到聚合物、晶体和生物分子溶液。
在详细的操作步骤前,先简单看一下在北鲲云超算平台提交Gromacs作业的方式。
一 模板提交
这是平台上最为简单的作业提交方式,只需找到软件,按照提示步骤上传作业,选择参数即可自动完成计算。大致界面如下。
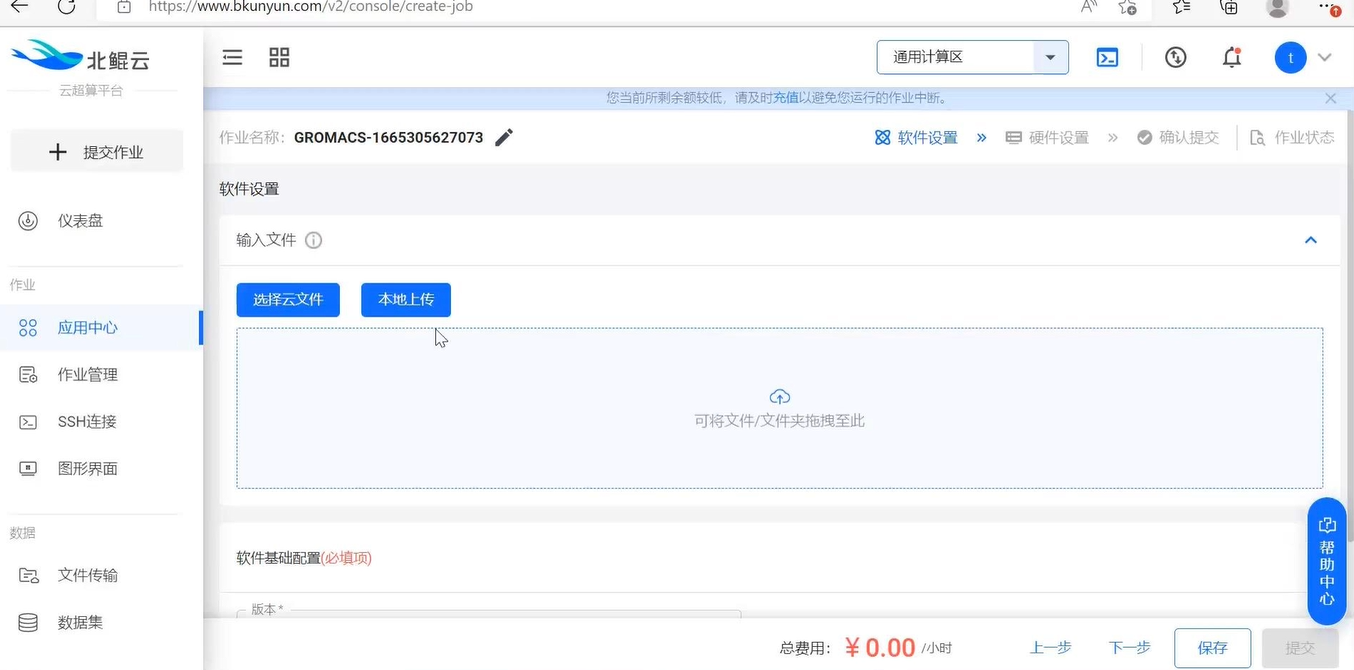
模板提交适合没有IT基础的用户,作业提交流程、配置参数全可视化界面。
二 命令行提交
命令行提交在管理节点使用SSH命令行创建动态集群,可以同时调用多种相同或者不同规格的计算节点,通过slurm作业调度系统提交作业计算,全程只需命令行操作。适合有一定代码基础的用户。
Step 1. 通过SSH连接创建并连接管理节点;
Step 2. 创建作业目录并进入;
mkdir gromacsJob1cd gromacsJob1
Step 3. 将运行gromacs需要的相关文件上传到该文件夹下,详情请查看Linux数据传输;
GPU版GROMACS作业示例
Step 1. 在该文件夹下创建如下执行脚本gromacs.sh:
#!/bin/bashmodule add GROMACS/2021-fosscuda-2019b #加载软件export GMX_GPU_DD_COMMS=true export GMX_GPU_PME_PP_COMMS=trueexport GMX_FORCE_UPDATE_DEFAULT_GPU=true#生成tpr格式输入文件,如果已有tpr格式文件则不需要写#gmx grompp -f pme.mdp -c conf.gro -p tpr_file.top -o tpr_file_name.tprgmx mdrun -v -pin on -nb gpu -bonded gpu -pme gpu -cpi tpr_file_name -deffnm tpr_file_name
Step 2. 使用slurm命令提交到计算节点;
sbatch -N 1 -p g-v100-1 -c 12 gromacs.sh
CPU版GROMACS作业示例
Step 1. 创建执行脚本:
#!/bin/bashmodule add GROMACS/2019.6-intel-2019b #加载软件mpiexec -v gmx_mpi mdrun -v -cpi tpr_file_name -deffnm tpr_file_name
Step 2. 提交作业;
2个4核心节点启动8个并行任务。
sbatch -N 2 -p c-4-1 -n 8 -c 1 gromacs.sh
查看作业运行情况及需使用slurm命令。具体可以查询北鲲云帮助手册。
以Gromacs在北鲲云超算平台完成溶菌酶在水中的动力学模拟为例
整个步骤分为10步
结构处理,文件上传,生成拓扑文件,定义单位盒子并填充溶剂,添加离子,体系能量最小化,NVT平衡,NPT平衡,动力学模拟成品模拟的部分可以到我们以前发布的文章中查看。
之前的教程非常详细的展示了如何用Gromacs进行动力学模拟,值得注意的是,
上述教程中的命令可以在单机完成,也可以上述所有命令写成作业脚本进行提交。提交脚本命令:
sbatch -N 1 -p g-v100-1 -c 12 md-gromacs.sh
其中,-N为节点的数量,这里输入的是1。-p为选择的PARTITION,这里使用的是V100卡(g-v100-1)。
md-gromacs.sh脚本的内容涵盖上述教程中的所有命令,根据北鲲云超算平台的指南需要在脚本开头加上导入gromacs模块,如果申请了GPU需要将GPU模块也导入(1-6行),具体脚本内容如下:
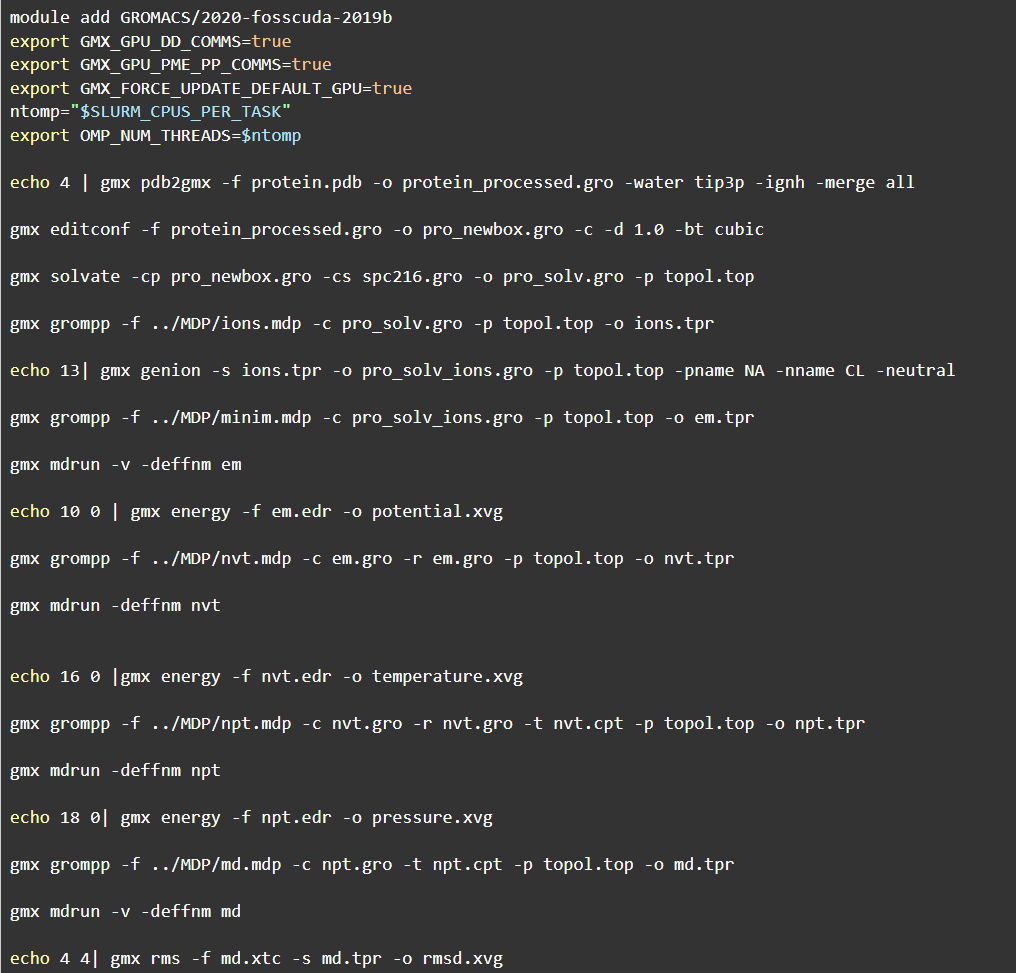
所有操作只需要可以登录北鲲云超算平台在线操作即可,无需自己配备高性能的计算机,和为繁琐的工具安装浪费时间。





















工程师必备
- 项目客服
- 培训客服
- 平台客服
TOP




